
СВЯЗЬ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ С БЕЛКАМИ ПЛАЗМЫ : ТОЧКА ЗРЕНИЯ АНЕСТЕЗИОЛОГА Margaret Wood, Vanderbilt Univercity , Nashville, TennesseeВлияние связывания препарата с белками на его распределение Именно свободная, или несвязанная фракция
препарата свободно проникает через мембраны,
таким образом, что равновесная концентрация в
межклеточной жидкости будет определяться именно
несвязанной формой препарата. В общем, чем больше
уровень связывания препарата с белками плазмы
крови, тем меньше лекарства может выйти за
пределы сосудов, и тем меньше будет объем его
распределения. Например, исследование отношения
между объемом распределения такого препарата,
как пропроанолол, и концентрацией свободной
фракции показывает, что объем распределения
повышается с увеличением свободной фракции.
Итак, частичный объем распределения является
показателем как распределения, так и связывания
препарата.
отсюда:
Но объем распределения в ткани равен:
Исходя из уравнения 3, он равен
Итак:
где Ср = общая концентрация в плазме, Ст= общая концентрация в ткани, Vd ss = изначальный объем распределения, Vp= объем плазмы, Vt= объем ткани, Fр= свободная фракция препарата в плазме и Ft = свободная фракция препарата в тканях.Итак, из уравнения 4, объем распределения зависит от связывания препарата как с белками крови, так и с белками плазмы. Для хорошо растворимых в липидах препаратов, которые в равной степени связываются как с плазмой, так и с тканями, и поэтому легко проникают через мембраны клеток, объем распределения свободной фракции будет равен количеству воды всего тела. Итак, если взять простейший пример препарата, который в равной мере связывается как плазмой, так и тканями, то мы получим следующее: fp = ft , отсюда fp/ft =1, Vdss = Vp + Vt = вода всего тела ( из уравнения 4). Для препаратов, которые не очень хорошо растворимы в липидах и поэтому с трудом проходят через мембраны, объем распределения будет равен объему всей межклеточной жидкости. Наименьший такой объем равен объему плазмы. Объем распределения, равный объему плазмы возможен только тогда, когда препарат так сильно связывается плазмой, что свободная фракция его в плазме близится к нулю, и тогда, когда его связывание в тканях равно нулю и, следовательно, свободная фракция в тканях приближается к 100 %. Итак, из уравнения 4, если fp --> 0, то Vd ss = Vp . Лекарства, которые хорошо связываются с белками плазмы, но не очень хорошо- с белками тканей ( fp/ft <1), имеют объем распределения больше объема плазмы, но меньше количества воды всего тела. Напротив, препараты, которые более интенсивно соединяются с белками тканей ( fp/ft >1), имеют объемы распределения, большие количества воды всего тела. Для препаратов, которые очень сильно связываются с белками тканей, свободная фракция их в ткани будет очень низкой, скажем, 1%, а свободная фракция их в плазме будет приближаться к 100%. Итак, если fp/ft>1, то Vdss = Vp + Vt [100/1] ( из уравнения 4). Однако, Vp + Vt примерно равны объему воды всего тела, и поэтому Vdss примерно в 100 раз превысит объем воды всего тела, что демонстрирует, что для препаратов, хорошо связывающихся с белками тканей, Vd больше объема воды всего тела. Антипирин, который минимально связывается как с белками плазмы, так и с белками тканей, и для которого и fp, и ft достигают 100%, имеет объем распределения, примерно равный объему воды всего тела ( fp/ft =1, Vdss = Vp +Vt ). Уровень связывания препарата с белками плазмы является важным фактором при распределении препарата между матерью и плодом, поскольку только несвязанная, то есть свободная фракция препарата может проходить через плаценту. Связывание с плазмой бупивакаина выше в плазме матери, чем в плазме у новорожденного (в связи с тем, что у новорожденного концентрация АГК ниже) и во время родов, после эпидуральной анестезии бупивакаином, общая концентрация бупивакаина в крови из пупочной вены будет ниже, чем в венозной крови матери. Бупивакаин хорошо связывается белками плазмы крови матери и поэтому только очень маленькая свободная фракция участвует в распределении и проходит через плаценту. Однако, свободная фракция бупивакаина в венозной крови матери и в крови из пупочной вены плода сразу после родов одинаковы. Поскольку только свободная форма может занять рецептор и оказать фармакологический эффект, эти эффекты будут одинаковы, несмотря на разницу общих концентраций бупивакаина в крови матери и плода. Это - хороший источник всяких конфузов, поскольку некоторые препараты считаются наиболее безопасными для использования в акушерской практике именно вследствие их более низкой общей концентрации в крови новорожденного, чем в крови матери, причем в этом случае никто не принимал во внимание различную связывающую способность плазмы крови матери и плода, что может очень серьезно повлиять на эффективную концентрацию препарата в крови новорожденного или плода. Время полуэлиминации препарата ( t 1/2) зависит как от объема его распределения (Vd), так и от клиренса препарата (Cl).Изменения связывания препарата с белками могут влиять как на объем распределения, так и на клиренс препарата, иногда в совершенно противоположных направлениях, поэтому очень трудно предсказать общий эффект изменений связываемости на время полужизни. Снижение связывания тиопентала с белками плазмы у больных с хронической почечной недостаточностью в результате вышеописанных причин не привело к изменению времени его полужизни, поскольку изменились как объем распределения, так и клиренс препарата. Все лекарственные препараты могут быть разделены на две группы в соответствии с той скоростью, с которой они удаляются, проходя через печень человека. Такие препараты, как пропранолол, морфин и лидокаин, которые быстро удаляются из крови при их прохождении через печень относятся к препаратам с высоким уровнем экстракции, тогда как препараты, плохо удаляемые печенью, к ним относятся варфарин и толбутамид, считаются препаратами низкого уровня экстракци. Их фармакокинетика всегда считалась зависящей от активности ферментов печени, тогда как кинетика препаратов высокого уровня экстракции зависит только от печеночного кровотока. Это может быть выражено следующим уравнением, которое пригодно для использования относительно любого органа: Клиренс (Cl)= Q * E, где Q = органный кровоток и Е= показатель экстракции. Влияние связывания препарата с белками на
печеночный клиренс зависит от показателя
экстракции препарата. Для препаратов с высоким
уровнем экстракции, для которых Е=1, клиренс
должен равняться кровотоку. Таким образом, для
них клиренс зависит практически только от
кровотока, поскольку весь препарат, который
проходит через орган, будет удален из крови, вне
зависимости от того, связан он с белком, или нет.
Например, показатель экстракции для
пропранолола составляет около 80% и пропранолол
хорошо связывается с белками: его свободная
фракция составляет 5-10 %. Изменения в связи
препарата с белками мало влияют на печеночный
клиренс пропранолола, поскольку показатель
экстракции в несколько раз больше величины
свободной фракции и поэтому препарат как бы
"отрывается" от белков плазмы при
прохождении его через печень. Большая часть
препарата, проходящего через печень, удаляется
вне зависимости от того, связан он с белками или
нет. Поэтому для препаратов с высокими
показателями экстракции печеночный клиренс
зависит только от кровотока и не зависит от
уровня их связывания с белками.
где f b = свободная фракция препарата в крови, Q= печеночный кровоток, Clfint = клиренс свободной фракции и ClH = печеночный клиренс.Для препаратов с высоким клиренсом тогда, когда значение Clint большое по сравнению со скоростью доставки препарата в печень, то клиренс будет практически равен кровотоку в печени (Clint>> Q и ClH = Q ) и значение свободной фракции практически не будет влиять на клиренс, до те пор, пока fb не станет очень маленькой, примерно 0.25%. Из общего для всех органов уравнения (Cl = Q*E),
можно сделать вывод, что уровень экскреции или
почечный клиренс тех препаратов, которые хорошо
выводятся почками, практически равен почечному
кровотоку и целиком зависит от него и
относительно нечувствительен к изменениям
связываемости препарата с белком. Напротив,
клиренс тех препаратов, которые плохо выводятся
почками, очень чувствителен к изменению их связи
с белками. где f u= свободная фракция препарата в плазме и GFR= cкорость клубочковой фильтрации.Поскольку альбумин не проходит в норме через мембрану клубочка, та часть препарата, которая связана с альбумином, не может быть профильтрована, и только свободная фракция проходит через почечный фильтр. Поэтому скорость клубочковой фильтрации препарата обратно пропорциональна уровню его связанности с белком. Препараты, которые выводятся преимущественно путем клубочковой фильтрации и к тому же хорошо связываются с белком, имеют очень длительное время полужизни. Только свободная фракция лекарственных препарата активно секретируются почечными канальцами. Для того, чтобы поддержать равновесие между свободной и связанной фракциями, снижение концентрации свободной фракции должно сопровождаться распадом комплекса белок-препарат. Аналогично описанному выше процессу элиминации в печени, этот процесс может быть достаточно быстрым - тогда показатель экскреции будет превышать величину свободной фракции, что указывает на то, что во время прохождения препарата через почки он "отрывается от белка". Примером такой ситуации может служить пеннициллин, который хорошо связывается с белками и очень быстро выводится почками в связи с очень высокой канальцевой секрецией и минимальной реабсорбцией. Поскольку его почечный клиренс нечувствителен к изменению связанности препарата с белками, то такой клиренс будет называться неограниченным. Почечный клиренс уменьшается, если к остальным процессам прибавляется процесс канальцевой реабсорбции. Интенсивная реабсорбция может так уменьшить коэффициент экстракции, что препарат попадет в группу длительно удаляемых. Если реабсорбируется какая- то постоянная часть препарата (FR) и секреции нет, то справедливо следующее уравнение: Это уравнение показывает,
что и в этом случае почечный клиренс прямо
пропорционален величине свободной фракции. Эффект связывания препаратов и фармакодинамика Хотя эффект связывания на фармакодинамику
уже давно и интенсивно изучался, до сих пор эта
область проблемы остается не известной. К
сожалению, действие связывания препаратов с
белками при назначении препарата, время
полужизни и клиренс изменяют отношение
концентрации препарата ко времени, и, таким
образом, изменяют фармакологический эффект в
данный конкретный период времени. Конкурентные взаимодействия и токсичность Известно, что лекарственные препараты
могут взаимодействовать друг с другом, при этом
могут наблюдаться нежелательные эффекты или
снижение терапевтической активности препаратов.
Назначаемые вместе препараты или эндогенные
вещества могут конкурировать за рецептор для
присоединения к белку, что выражается в
конкурентных взаимодействиях. Снижение
связывания препарата с белком или его вытеснение
с места связывания могут усилить действие
препарата, однако, поскольку обычно несколько
таких процессов проходят параллельно, то
результат может быть непредсказуемым.
Одновременное назначение второго препарата
может вызвать вытеснение первого с рецептора на
белке, но концентрация этого второго препарата
должна обязательно достигать молярной
концентрации мест для связывания, а иначе
конкуренция будет невозможной. Любое
энодогенное или экзогенное вещество, связывание
которого зависит от концентрации (увеличение
свободной фракции с увеличением общей
концентрации) в терапевтических концентрациях
может вызвать такие процессы . Примерами таких
препаратов являются фенилбутазон и
сульфаниламиды. Все врачи понимают, что практически для
всех препаратов существует тесная взаимосвязь
между их общей концентрацией в плазме и эффектом.
Однако, для тех препаратов, которые хорошо
связываются с белками плазмы крови, эффект (будь
он терапевтическим или токсическим) может
принципиально зависеть от величины свободной
фракции препарата в плазме крови , таким образом,
понимание факторов, влияющих на связывание
препарата с белками плазмы крайне важно для
грамотной интерпретации взаимоотношения
эффекта и концентрации препарата. Приложение Связывание с белками плазмы крови препаратов, наиболее часто применяемых в анестезиологии
Anesthesia and analgesia
|
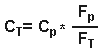 (3)
(3)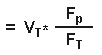
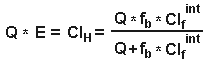 (7),
(7),